
Simprot Introduction
Protein evolution has been largely modeled by considering the amino
acid substitution process; however there have been few studies of the process
of insertion and deletion.
Simprot allows for several models of amino acid substitution (PAM,
JTT and PMB), gamma distributed sites rates according to Yang's model,
and implements a parameterized Qian and Goldstein distribution model for
insertion and deletion. Also it is possible to use Zipfian's distribution
of indels in proteins.
For comments, problems and doubts please send an email message to Elisabeth
Tillier
Implementation
Simprot is a cross-platform software written in C++.
The program uses a bifurcating Newick phylogenetic trees as input. The
result of the simulation is output in three different files:
- an alignment file, fasta format (TXT extension)
- a sequence file, fasta format (SEQ.TXT extension)
- a Phylip file, phylip format (TXT.PHY extension)
The simulation parameters that can be set are:
- Branch length scale multiplier (default: 1)
- Root length (default: 50), which is the sequence size to be simulated
- Evolutionary Scale factor (default: 3, when Qian-Goldstein is selected),
for the distribution of indel lengths
- Indel frequency (default: 0.03) for evolutionary time c
- Maximum insertion/deletion length (default: 2048). This limit is beyond
the 20% length of the sequence limit.
- Amino acid substitution model: PAM, JTT and PMB (default)
- variable gamma on insertions (default: false). When true the average
rate of evolution for insertions it the rate of the amino acid where the
insertion has occurred.
- Qian and Goldstein or Zipfian distributions (default: Qian and Goldstein)
- Parameter theta of Zipfian distribution (default: -2, when Zipfian
is selected), used on the distribution of indel lengths
- Gamma alpha (default: 1)
Notes:
The evolutionary rate is normalized to an average of 1 over the whole
protein
When Zipfian distribution model is selected, the frequency of indels
(p) is determined with PAM = 100.
There are strict limits on the
size of the tree, sequences and number of simulations that can be run from
the web server. Use local installation if you are having problems.
The default parameters are supplied by the program as soon as it starts.
Local installations of Simprot can generate single or multiple segments
simulations, using different tree files and parameters for each run.
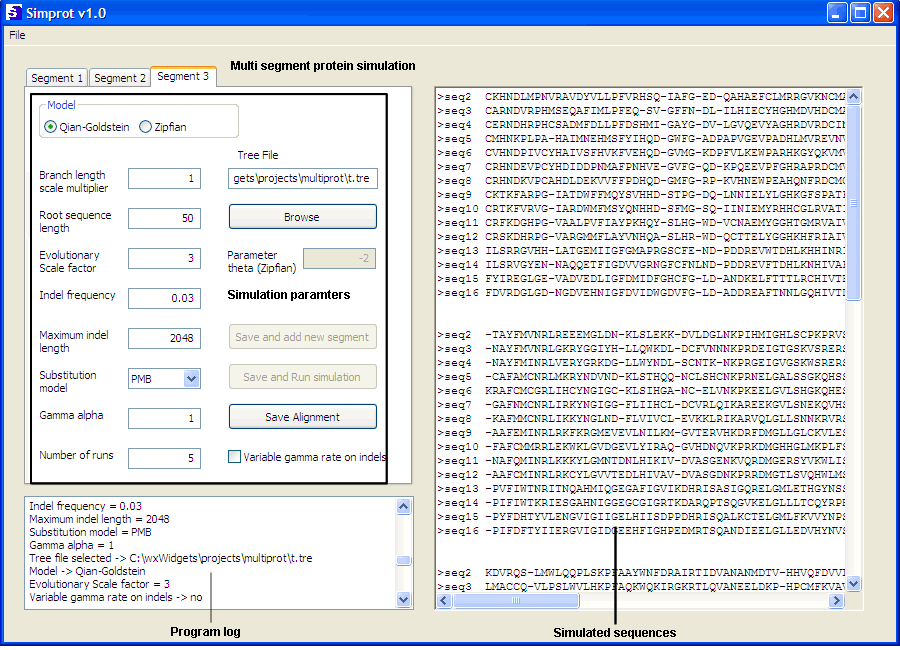
Simprot tutorial
Single segment simulation
1- Modify the parameters with the desired values in the respective text
boxes, radio box and check box. Number of runs determine the number
of alignments to be generated.
2- Click on Browse and select the tree file in the file open
dialog.
3- Click on Save and Run simulation to start. The log is updated
with the entered values and the simulation being executed.
4- The final alignment(s) will be displayed on the large text box on
the right.
5- To save the result, click on Save Alignment and select the
path and file name to be saved. Click OK and Simprot generates the 3 resulting
files.
Multiple segments simulation
1- Modify the parameters with the desired values in the respective text
boxes, radio box and check box. Number of runs determine the number
of alignments to be generated (Note: Simprot only takes in account the
number of runs entered in the last segment to be simulated).
2- Click on Browse and select the tree file in the file open
dialog.
3- Click on Save and add segment. Repeat steps 1 and 2.
4- When the last segment is set click on Save and Run simulation
to start. The log is updated with the entered values and the simulation
being executed.
5- The final alignment(s) will be displayed on the big text box on the
right.
6- To save the result, click on Save Alignment and select the
path and file name to be saved. Click OK and Simprot generates the 3 resulting
files.
/* Simprot (c) Copyright 2005 by the University Health Network written
by Elisabeth Tillier.
Permission is granted to copy and use this program provided no fee
is charged for it
and provided that this copyright notice is not removed. */